Chapter 3 RNA Data preprocessing
3.1 Introduction
Transcriptomic data primarily encompasses microarray data and RNA-seq data. Microarray data are predominantly derived from platforms such as Affymetrix and Illumina, while RNA-seq data are largely generated using second-generation sequencing technologies, with third-generation sequencing becoming increasingly available. When performing TME analysis and calculating signature scores, it is essential to first annotate and normalize the gene expression matrix.
The IOBR package offers a suite of functions designed to facilitate rapid preprocessing of transcriptomic data, including:
- count2tpm: Performs gene annotation, removes duplicate genes, and applies TPM normalization.
- anno_eset: Handles duplicate gene removal and gene annotation.
- find_outlier_samples: Identifies and removes outlier samples.
- iobr_pca: Visualizes batch effects and examines biological variability.
- remove_batcheffect: Removes batch effects and integrates multiple datasets.
Streamlining Gene Annotation for Downstream Analysis
Since downstream TME analysis tools and scoring functions in IOBR primarily recognize gene symbols rather than probe IDs, Ensembl IDs, or Entrez IDs, the anno_eset() function simplifies the annotation process. It enables efficient annotation of microarray data (e.g., Affymetrix and Illumina) and RNA-seq data (e.g., Ensembl and Entrez IDs), converting these identifiers into the more familiar gene symbols.
Improving Inter-Sample Comparability Through Normalization
In RNA-seq data analysis, TPM (transcripts per million) is a widely adopted normalization method that converts raw read counts into relative abundance or expression levels. TPM normalization accounts not only for the read count of each gene but also for the gene length, providing a more accurate comparison of expression levels across genes. This method effectively addresses the bias introduced by varying gene lengths, improving the comparability of expression levels between different genes.
IOBR offers the count2tpm() function for TPM conversion of RNA-seq count data. For microarray data, which is typically pre-normalized, packages such as affy and limma are commonly used for preprocessing. However, due to the higher cost and relatively limited information content of microarrays, RNA-seq has become the dominant technology in transcriptomic studies.
3.3 Gene Annotation
Annotation of genes in the expression matrix and removal of duplicate genes.
# Load the annotation file `anno_hug133plus2` in IOBR.
anno_hug133plus2 <- load_data("anno_hug133plus2")
head(anno_hug133plus2)## # A tibble: 6 × 2
## probe_id symbol
## <fct> <fct>
## 1 1007_s_at MIR4640
## 2 1053_at RFC2
## 3 117_at HSPA6
## 4 121_at PAX8
## 5 1255_g_at GUCA1A
## 6 1294_at MIR5193# Load the annotation file `anno_grch38` in IOBR.
anno_grch38 <- load_data("anno_grch38")
head(anno_grch38)## id eff_length gc entrez symbol chr start end
## 1 ENSG00000000003 4536 0.3992504 7105 TSPAN6 X 100627109 100639991
## 2 ENSG00000000005 1476 0.4241192 64102 TNMD X 100584802 100599885
## 3 ENSG00000000419 9276 0.4252911 8813 DPM1 20 50934867 50958555
## 4 ENSG00000000457 6883 0.4117391 57147 SCYL3 1 169849631 169894267
## 5 ENSG00000000460 5970 0.4298157 55732 C1orf112 1 169662007 169854080
## 6 ENSG00000000938 3382 0.5644589 2268 FGR 1 27612064 27635277
## strand biotype
## 1 -1 protein_coding
## 2 1 protein_coding
## 3 -1 protein_coding
## 4 -1 protein_coding
## 5 1 protein_coding
## 6 -1 protein_coding
## description
## 1 tetraspanin 6 [Source:HGNC Symbol;Acc:HGNC:11858]
## 2 tenomodulin [Source:HGNC Symbol;Acc:HGNC:17757]
## 3 dolichyl-phosphate mannosyltransferase polypeptide 1, catalytic subunit [Source:HGNC Symbol;Acc:HGNC:3005]
## 4 SCY1-like, kinase-like 3 [Source:HGNC Symbol;Acc:HGNC:19285]
## 5 chromosome 1 open reading frame 112 [Source:HGNC Symbol;Acc:HGNC:25565]
## 6 FGR proto-oncogene, Src family tyrosine kinase [Source:HGNC Symbol;Acc:HGNC:3697]# Load the annotation file `anno_gc_vm32` in IOBR for mouse RNAseq data
anno_gc_vm32 <- load_data("anno_gc_vm32")
head(anno_gc_vm32)## id eff_length gc symbol mgi_id gene_type
## 1 ENSMUSG00000000001 3262 0.4350092 Gnai3 MGI:95773 protein_coding
## 2 ENSMUSG00000000003 902 0.3481153 Pbsn MGI:1860484 protein_coding
## 3 ENSMUSG00000000028 3506 0.4962921 Cdc45 MGI:1338073 protein_coding
## 4 ENSMUSG00000000031 2625 0.5588571 H19 MGI:95891 lncRNA
## 5 ENSMUSG00000000037 6397 0.4377052 Scml2 MGI:1340042 protein_coding
## 6 ENSMUSG00000000049 1594 0.5050188 Apoh MGI:88058 protein_coding
## start end transcript_id ont
## 1 108014596 108053462 <NA> <NA>
## 2 76881507 76897229 <NA> <NA>
## 3 18599197 18630737 <NA> <NA>
## 4 142129262 142131886 <NA> <NA>
## 5 159865521 160041209 <NA> <NA>
## 6 108234180 108305222 <NA> <NA>3.4 Download array data using GEOquery
Obtaining data set from GEO Gastric cancer: GSE62254 using GEOquery R package.
if (!requireNamespace("GEOquery", quietly = TRUE)) BiocManager::install("GEOquery")
library("GEOquery")
# NOTE: This process may take a few minutes which depends on the internet connection speed. Please wait for its completion.
eset_geo <- getGEO(GEO = "GSE62254", getGPL = F, destdir = "./")
eset <- eset_geo[[1]]
eset <- exprs(eset)
eset[1:5, 1:5]## GSM1523727 GSM1523728 GSM1523729 GSM1523744 GSM1523745
## 1007_s_at 3.2176645 3.0624323 3.0279131 2.921683 2.8456013
## 1053_at 2.4050109 2.4394879 2.2442708 2.345916 2.4328582
## 117_at 1.4933412 1.8067380 1.5959665 1.839822 1.8326058
## 121_at 2.1965561 2.2812181 2.1865556 2.258599 2.1874363
## 1255_g_at 0.8698382 0.9502466 0.8125414 1.012860 0.94419933.4.1 For Array data: HGU133PLUS-2 (Affaymetrix)
# Conduct gene annotation using `anno_hug133plus2` file; If identical gene symbols exists, these genes would be ordered by the mean expression levels. The gene symbol with highest mean expression level is selected and remove others.
eset <- anno_eset(
eset = eset,
annotation = anno_hug133plus2,
symbol = "symbol",
probe = "probe_id",
method = "mean"
)
eset[1:5, 1:3]## GSM1523727 GSM1523728 GSM1523729
## SH3KBP1 4.327974 4.316195 4.351425
## RPL41 4.246149 4.246808 4.257940
## EEF1A1 4.293762 4.291038 4.262199
## COX2 4.250288 4.283714 4.270508
## LOC101928826 4.219303 4.219670 4.2132523.5 Download RNAseq data using UCSCXenaTools
In this section, we are going to download RNA-seq data from The Cancer Genome Atlas (TCGA) for applying the downstream analysis workflow of IOBR. Particularly, we will use the convenient R package UCSCXenaTools to query and download the RNA-seq data of TCGA stomach cancer cohort.
Use the following code to check and install UCSCXenaTools.
if (!requireNamespace("UCSCXenaTools", quietly = TRUE)) {
BiocManager::install("ropensci/UCSCXenaTools")
}UCSCXenaTools provides an R interface to access public cancer datasets from UCSC Xena data hubs, including multiple pan-cancer studies like TCGA and PCAWG. You can directly access information of all datasets in R.
## =========================================================================================
## UCSCXenaTools version 1.7.0
## Project URL: https://github.com/ropensci/UCSCXenaTools
## Usages: https://cran.r-project.org/web/packages/UCSCXenaTools/vignettes/USCSXenaTools.html
##
## If you use it in published research, please cite:
## Wang et al., (2019). The UCSCXenaTools R package: a toolkit for accessing genomics data
## from UCSC Xena platform, from cancer multi-omics to single-cell RNA-seq.
## Journal of Open Source Software, 4(40), 1627, https://doi.org/10.21105/joss.01627
## =========================================================================================
## --Enjoy it--##
## Attaching package: 'UCSCXenaTools'## The following object is masked from 'package:Biobase':
##
## samples## # A tibble: 6 × 17
## XenaHosts XenaHostNames XenaCohorts XenaDatasets SampleCount DataSubtype Label
## <chr> <chr> <chr> <chr> <int> <chr> <chr>
## 1 https://… publicHub Breast Can… ucsfNeve_pu… 51 gene expre… Neve…
## 2 https://… publicHub Breast Can… ucsfNeve_pu… 57 phenotype Phen…
## 3 https://… publicHub Glioma (Ko… kotliarov20… 194 copy number Kotl…
## 4 https://… publicHub Glioma (Ko… kotliarov20… 194 phenotype Phen…
## 5 https://… publicHub Lung Cance… weir2007_pu… 383 copy number CGH
## 6 https://… publicHub Lung Cance… weir2007_pu… 383 phenotype Phen…
## # ℹ 10 more variables: Type <chr>, AnatomicalOrigin <chr>, SampleType <chr>,
## # Tags <chr>, ProbeMap <chr>, LongTitle <chr>, Citation <chr>, Version <chr>,
## # Unit <chr>, Platform <chr>UCSCXenaTools provides workflow functions to generate object, filter, query, download and load the dataset(s) of interest. The following code show a standardized UCSCXenaTools data workflow to query the data from UCSC Xena data hub and load it into R.
library(UCSCXenaTools)
# NOTE: This process may take a few minutes which depends on the internet connection speed. Please wait for its completion.
eset_stad <- XenaGenerate(subset = XenaCohorts == "GDC TCGA Stomach Cancer (STAD)") %>%
XenaFilter(filterDatasets = "TCGA-STAD.star_counts.tsv") %>%
XenaQuery() %>%
XenaDownload() %>%
XenaPrepare()
eset_stad[1:5, 1:3]As the metadata of this dataset have been stored in the XeneData data.frame. You can easily recheck the dataset with code.
3.6 Normalization and Gene annotation
Transform gene expression matrix into TPM format, and conduct subsequent annotation.
# Remove Ensembl IDs with the suffix '_PAR_Y'.
eset_stad <- eset_stad[!grepl("_PAR_Y$", eset_stad$Ensembl_ID), ]
# Remove the version numbers in Ensembl ID.
eset_stad$Ensembl_ID <- substring(eset_stad$Ensembl_ID, 1, 15)
eset_stad <- column_to_rownames(eset_stad, var = "Ensembl_ID")
# Revert back to original format because the data from UCSC was log2(x+1)transformed.
eset_stad <- (2^eset_stad) - 1
eset_stad <- count2tpm(countMat = eset_stad, idType = "Ensembl", org = "hsa", source = "local")
eset_stad[1:5, 1:5]3.7 Identifying outlier samples
Take ACRG microarray data for example
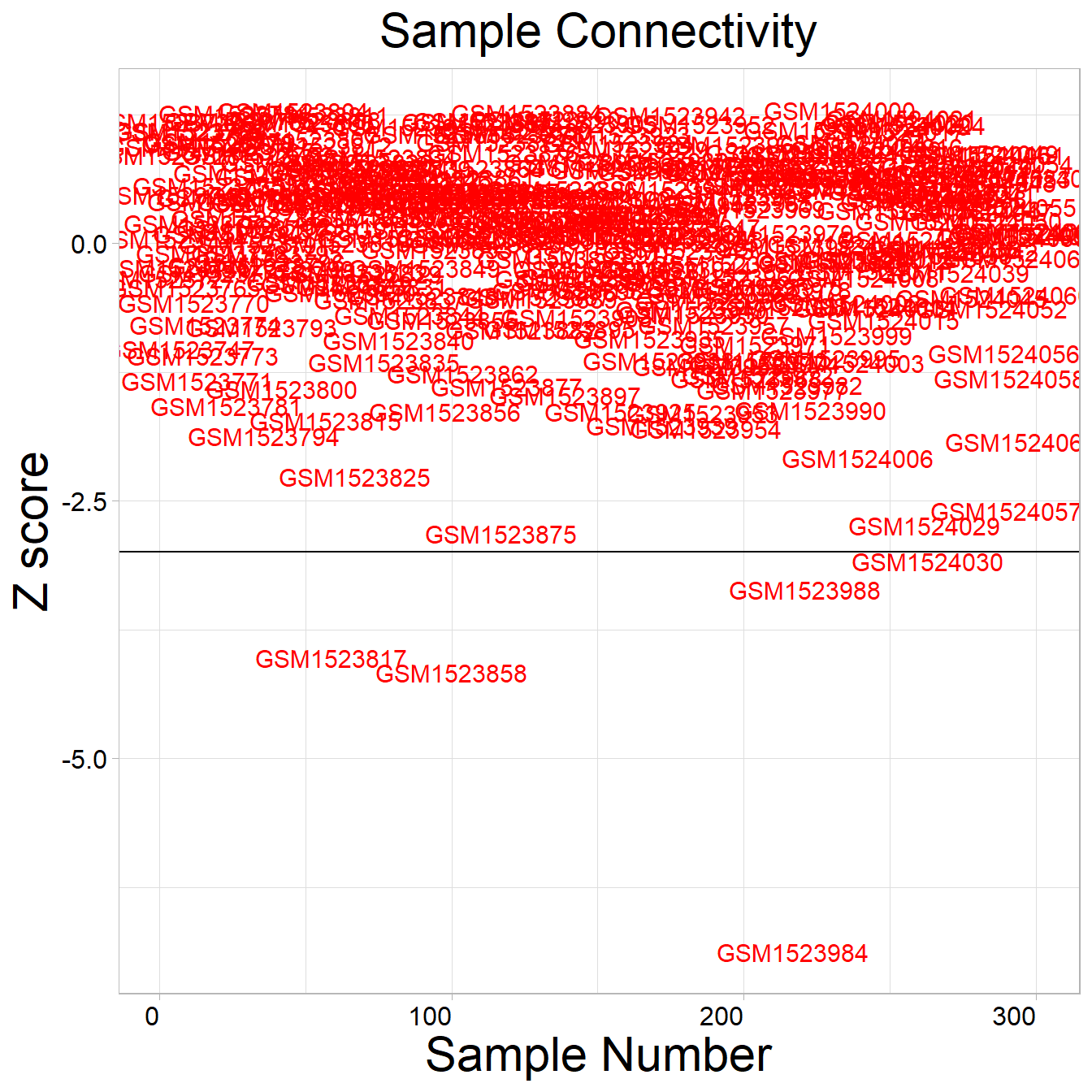
Removing potential outlier samples
3.8 PCA analysis of molecular subtypes
pdata_acrg <- load_data("pdata_acrg")
res <- iobr_pca(
data = eset1,
is.matrix = TRUE,
scale = TRUE,
is.log = FALSE,
pdata = pdata_acrg,
id_pdata = "ID",
group = "Subtype",
geom.ind = "point",
cols = "normal",
palette = "jama",
repel = FALSE,
ncp = 5,
axes = c(1, 2),
addEllipses = TRUE
)##
## CIN EBV EMT GS MSI MSS/TP53- MSS/TP53+
## 0 0 42 0 68 106 79## [1] ">>== colors for group: "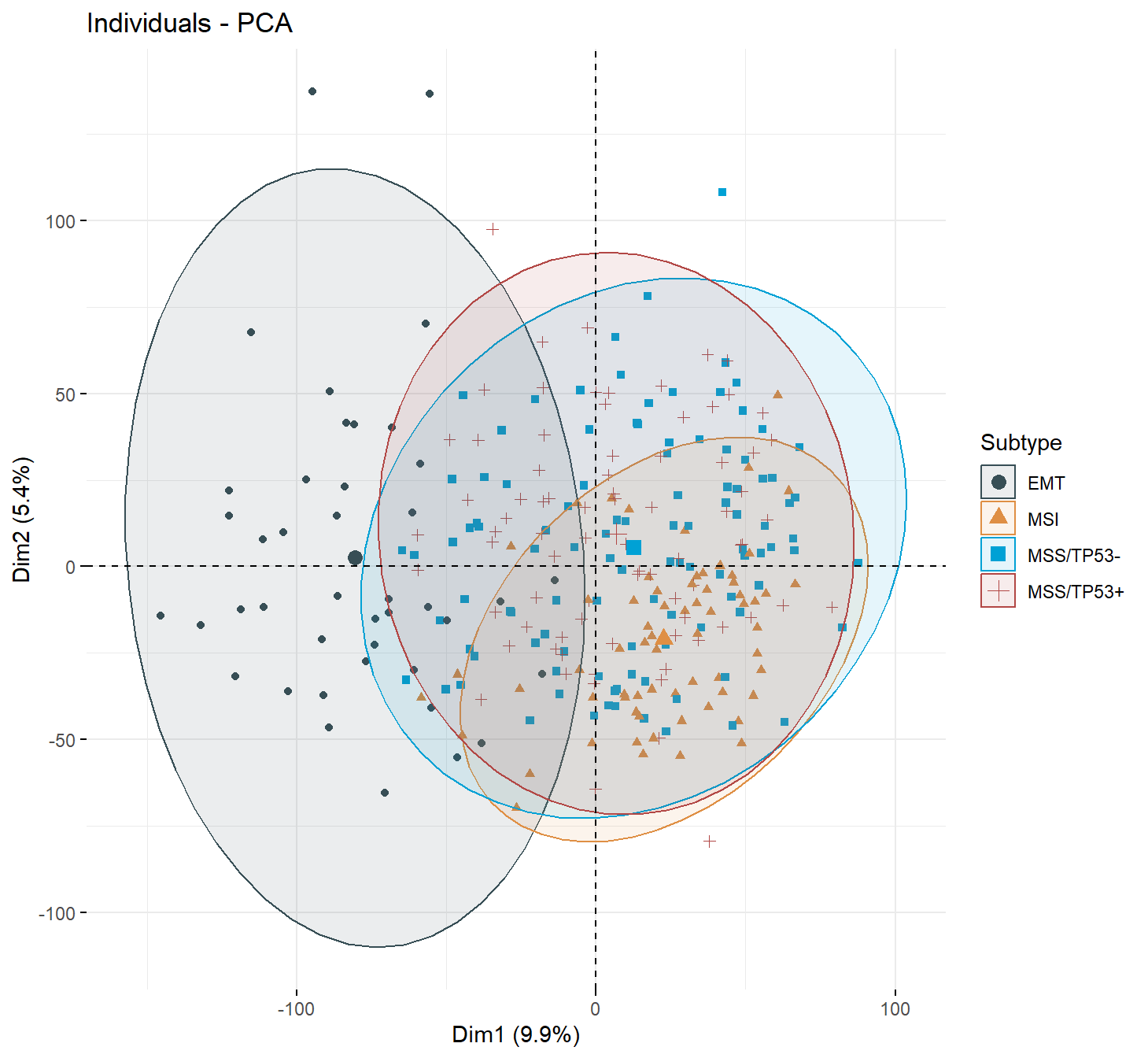
3.9 Batch effect correction
3.9.1 For microarray data
Obtaining another data set from GEO Gastric cancer: GSE57303 using GEOquery R package.
# NOTE: This process may take a few minutes which depends on the internet connection speed. Please wait for its completion.
eset_geo <- getGEO(GEO = "GSE57303", getGPL = F, destdir = "./")
eset2 <- eset_geo[[1]]
eset2 <- exprs(eset2)
eset2[1:5, 1:5]## GSM1379261 GSM1379262 GSM1379263 GSM1379264 GSM1379265
## 1007_s_at 8.34746 9.67994 8.62643 8.59301 8.63046
## 1053_at 5.07972 4.46377 5.29685 5.78983 4.33359
## 117_at 5.65558 4.48732 4.21615 5.47984 5.20816
## 121_at 5.95123 7.09056 6.19903 5.89872 5.91323
## 1255_g_at 1.66923 1.98758 1.73083 1.56687 1.63332Annotation of genes in the expression matrix and removal of duplicate genes.
eset2 <- anno_eset(
eset = eset2,
annotation = anno_hug133plus2,
symbol = "symbol",
probe = "probe_id",
method = "mean"
)
eset2[1:5, 1:5]## GSM1379261 GSM1379262 GSM1379263 GSM1379264 GSM1379265
## ND4 13.1695 13.1804 13.0600 12.4544 13.0457
## ATP6 13.1433 13.0814 13.0502 12.4831 13.1168
## SH3KBP1 12.9390 13.1620 12.9773 12.8745 13.1169
## COX2 13.0184 13.0489 12.8621 12.7489 12.9732
## RPL41 13.0201 12.6034 12.7929 13.0153 12.9404eset_com <- remove_batcheffect(
eset1 = eset1,
eset2 = eset2,
eset3 = NULL,
id_type = "symbol",
data_type = "array",
cols = "normal",
palette = "jama",
log2 = TRUE,
check_eset = TRUE,
adjust_eset = TRUE,
repel = FALSE,
path = "result"
)##
## eset1 eset2
## 295 70## [1] ">>== colors for group: "##
## eset1 eset2
## 295 70## [1] ">>== colors for group: "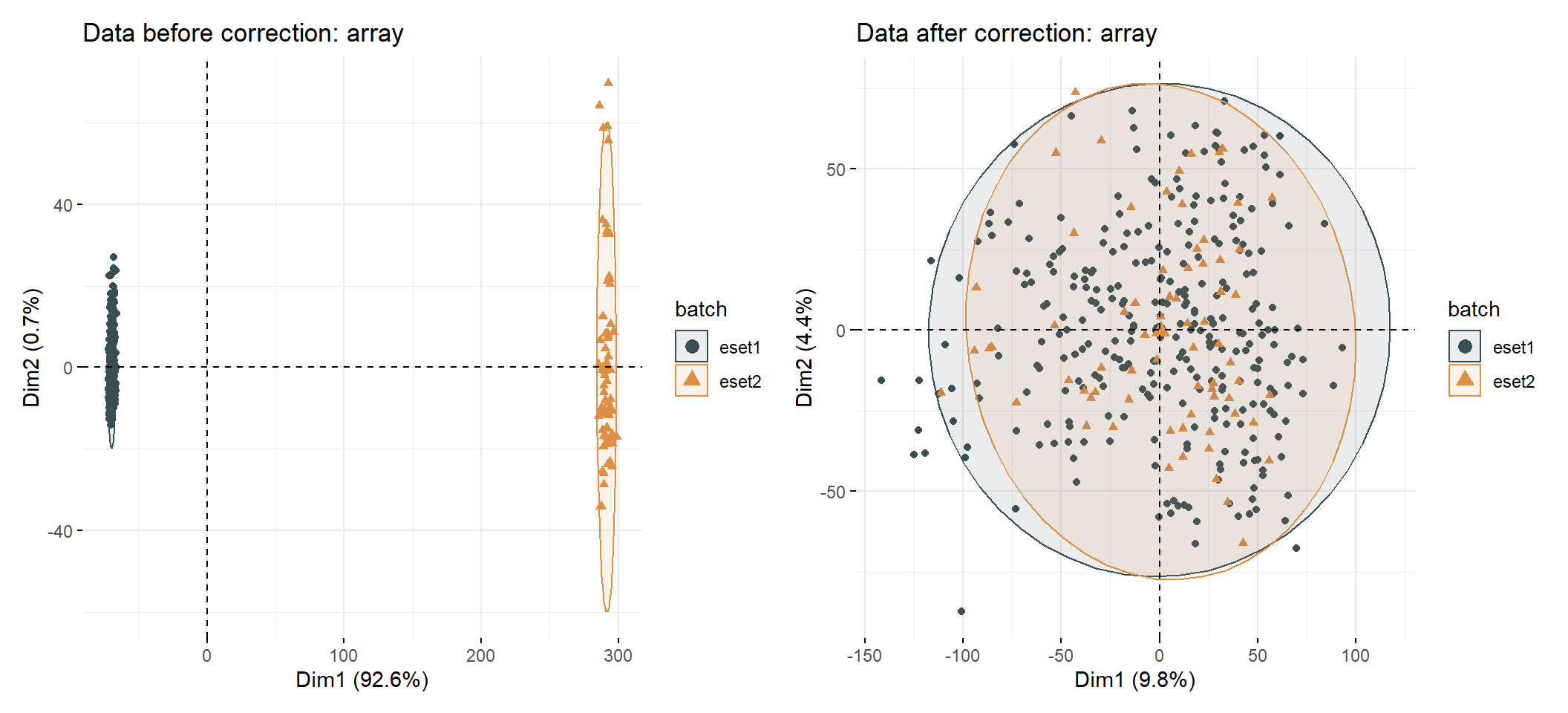
## [1] 21752 3653.9.2 For RNAseq count data
## TCGA-BR-6455 TCGA-BR-7196 TCGA-BR-8371 TCGA-BR-8380
## ENSG00000000003 8006 2114 767 1556
## ENSG00000000005 1 0 5 5
## ENSG00000000419 3831 2600 1729 1760
## ENSG00000000457 1126 745 1040 1260
## ENSG00000000460 857 463 231 432
## ENSG00000000938 758 1126 557 557
## TCGA-BR-8592 TCGA-BR-8686 TCGA-BR-A4IV TCGA-BR-A4J4
## ENSG00000000003 2806 2923 1524 7208
## ENSG00000000005 60 1 22 2
## ENSG00000000419 2273 1934 2838 4418
## ENSG00000000457 1814 707 1683 1335
## ENSG00000000460 635 323 270 423
## ENSG00000000938 828 666 760 597
## TCGA-BR-A4J9 TCGA-FP-7916
## ENSG00000000003 711 2747
## ENSG00000000005 0 3
## ENSG00000000419 2426 2824
## ENSG00000000457 1590 1672
## ENSG00000000460 276 773
## ENSG00000000938 370 688## TCGA-2F-A9KO TCGA-2F-A9KP TCGA-2F-A9KQ TCGA-2F-A9KR
## ENSG00000000003 6092 11652 5426 4383
## ENSG00000000005 0 4 1 1
## ENSG00000000419 3072 2656 1983 2061
## ENSG00000000457 1302 984 1134 1092
## ENSG00000000460 779 924 421 386
## ENSG00000000938 436 116 312 590
## TCGA-2F-A9KT
## ENSG00000000003 3334
## ENSG00000000005 0
## ENSG00000000419 2930
## ENSG00000000457 496
## ENSG00000000460 318
## ENSG00000000938 362## Found 2 batches
## Using null model in ComBat-seq.
## Adjusting for 0 covariate(s) or covariate level(s)
## Estimating dispersions
## Fitting the GLM model
## Shrinkage off - using GLM estimates for parameters
## Adjusting the data##
## eset1 eset2
## 10 5## [1] ">>== colors for group: "##
## eset1 eset2
## 10 5## [1] ">>== colors for group: "##
## eset1 eset2
## 10 5## [1] ">>== colors for group: "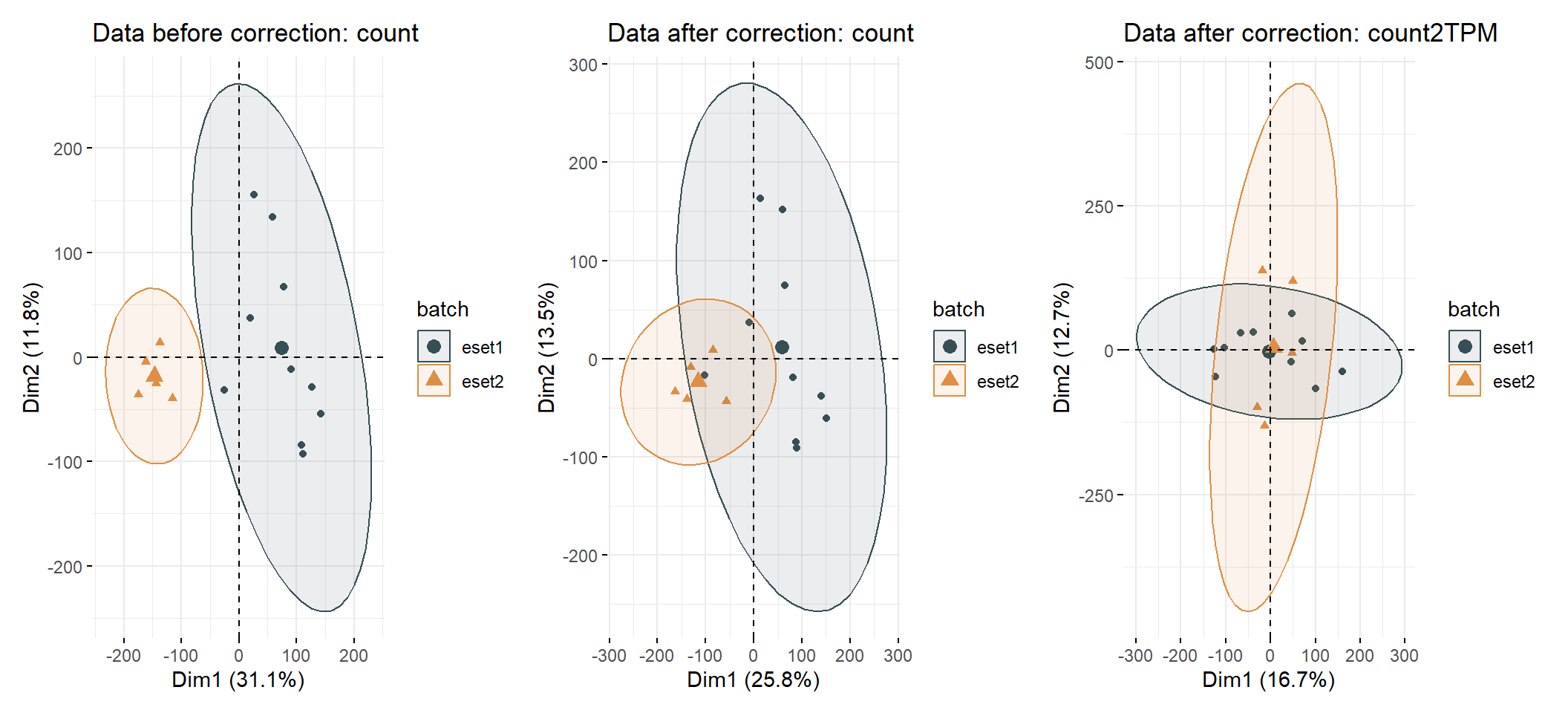
## TCGA-BR-6455 TCGA-BR-7196 TCGA-BR-8371 TCGA-BR-8380
## ENSG00000000003 10264 3536 1710 2964
## ENSG00000000005 1 0 4 5
## ENSG00000000419 4500 3099 2111 2167
## ENSG00000000457 1203 707 1106 1353
## ENSG00000000460 1059 590 310 560
## ENSG00000000938 731 1202 507 485
## TCGA-BR-8592 TCGA-BR-8686 TCGA-BR-A4IV TCGA-BR-A4J4
## ENSG00000000003 4761 3964 3115 9565
## ENSG00000000005 33 1 14 3
## ENSG00000000419 2782 2270 3444 5176
## ENSG00000000457 2089 817 1845 1469
## ENSG00000000460 810 405 368 548
## ENSG00000000938 769 723 677 532
## TCGA-BR-A4J9 TCGA-FP-7916 TCGA-2F-A9KO TCGA-2F-A9KP
## ENSG00000000003 1739 4371 2812 6796
## ENSG00000000005 0 3 0 10
## ENSG00000000419 2943 3362 2189 1849
## ENSG00000000457 1804 2044 994 817
## ENSG00000000460 371 959 495 584
## ENSG00000000938 281 654 456 156
## TCGA-2F-A9KQ TCGA-2F-A9KR TCGA-2F-A9KT
## ENSG00000000003 1971 1429 1057
## ENSG00000000005 1 1 0
## ENSG00000000419 1355 1420 2094
## ENSG00000000457 916 876 438
## ENSG00000000460 251 230 190
## ENSG00000000938 353 604 3833.10 References
Wang et al., (2019). The UCSCXenaTools R package: a toolkit for accessing genomics data from UCSC Xena platform, from cancer multi-omics to single-cell RNA-seq. Journal of Open Source Software, 4(40), 1627, https://doi.org/10.21105/joss.01627
Zhang et al., ComBat-seq: batch effect adjustment for RNA-seq count data, NAR Genomics and Bioinformatics, Volume 2, Issue 3, September 2020, lqaa078, https://doi.org/10.1093/nargab/lqaa078
Leek, J. T., et al., (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics, 28(6), 882-883.