Chapter 9 TME and genomic interaction
9.2 Genomic data prepare
In this section, we are going to use the MAF data of TCGA-STAD cohort as an example dataset. This dataset could be found in multiple places, here we show two ways to get it.
9.2.1 Using TCGAbiolinks to download genomic data
R Bioconductor package TCGAbiolinks provides an R interface of GDC data portal, which stores updating TCGA data. You can check and install this package with the following code.
Then you can query, download and prepare the required dataset.
library(TCGAbiolinks)
query <- GDCquery(
project = "TCGA-STAD",
data.category = "Simple Nucleotide Variation",
access = "open",
data.type = "Masked Somatic Mutation",
workflow.type = "Aliquot Ensemble Somatic Variant Merging and Masking"
)
GDCdownload(query)
maf <- GDCprepare(query)This maf object is a data.frame, you can use read.maf() from R package Maftools to convert it to a MAF object.
In this example, we used the maf file of TCGA-STAD to extract the SNPs in it,and then transformed it into a non-negative matrix.
## [1] "maf"## -Validating
## -Silent variants: 45460
## -Summarizing
## --Possible FLAGS among top ten genes:
## TTN
## MUC16
## SYNE1
## FLG
## -Processing clinical data
## --Missing clinical data
## -Finished in 6.166s elapsed (6.172s cpu)## Frame_Shift_Del Frame_Shift_Ins In_Frame_Del
## 21547 4526 1196
## In_Frame_Ins Missense_Mutation Nonsense_Mutation
## 106 102137 5669
## Nonstop_Mutation Splice_Site Translation_Start_Site
## 117 2242 107## DEL INS ONP SNP
## 22997 4675 10 1099659.3 Identifying Mutations Associated with TME
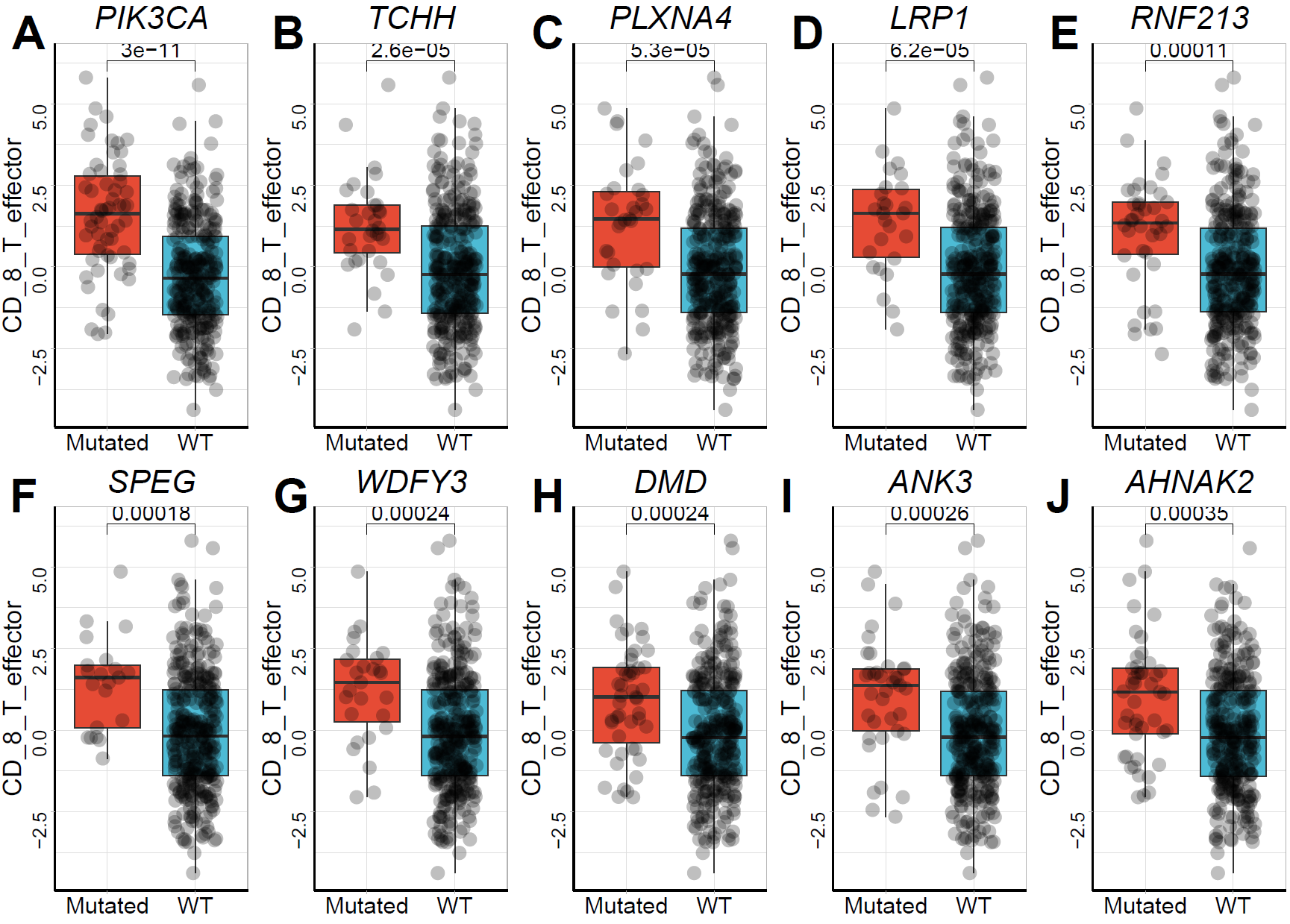
The microenvironmental data from the TCGA-STAD expression matrix was merged. The Cuzick or Wilcoxon test was used to identify genetic variants associated with microenvironmental factors. CD_8_T_effector was used as the target variable in this example.
## ℹ Loading cached data: "tcga_stad_sig"res <- tryCatch(
{
find_mutations(
mutation_matrix = mut,
signature_matrix = tcga_stad_sig,
id_signature_matrix = "ID",
signature = "CD_8_T_effector",
palette = "nrc",
min_mut_freq = 0.01,
plot = TRUE,
jitter = TRUE
)
},
error = function(e) {
message("find_mutations error: ", e$message)
NULL
}
)## Warning: The `size` argument of `element_line()` is deprecated as of ggplot2 3.4.0.
## ℹ Please use the `linewidth` argument instead.
## ℹ The deprecated feature was likely used in the IOBR package.
## Please report the issue at <https://github.com/IOBR/IOBR/issues>.
## This warning is displayed once per session.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
## generated.## [1] ">>>> Result of Cuzick Test"
## p.value names statistic adjust_pvalue
## PIK3CA 6.279087e-10 PIK3CA 6.183261 3.139543e-07
## PLXNA4 8.196539e-05 PLXNA4 3.938580 2.049135e-02
## DMD 3.514677e-04 DMD 3.574075 3.882570e-02
## SPEG 3.665757e-04 SPEG 3.563047 3.882570e-02
## AHNAK2 3.882570e-04 AHNAK2 3.547940 3.882570e-02
## TCHH 6.571311e-04 TCHH 3.406867 5.476093e-02
## ABCC9 1.043516e-03 ABCC9 3.278524 7.453684e-02
## ANK3 1.470480e-03 ANK3 3.180447 7.713409e-02
## WDFY3 1.690248e-03 WDFY3 3.139867 7.713409e-02
## LRP1 1.762002e-03 LRP1 3.127666 7.713409e-02## find_mutations error: object 'file_name' not found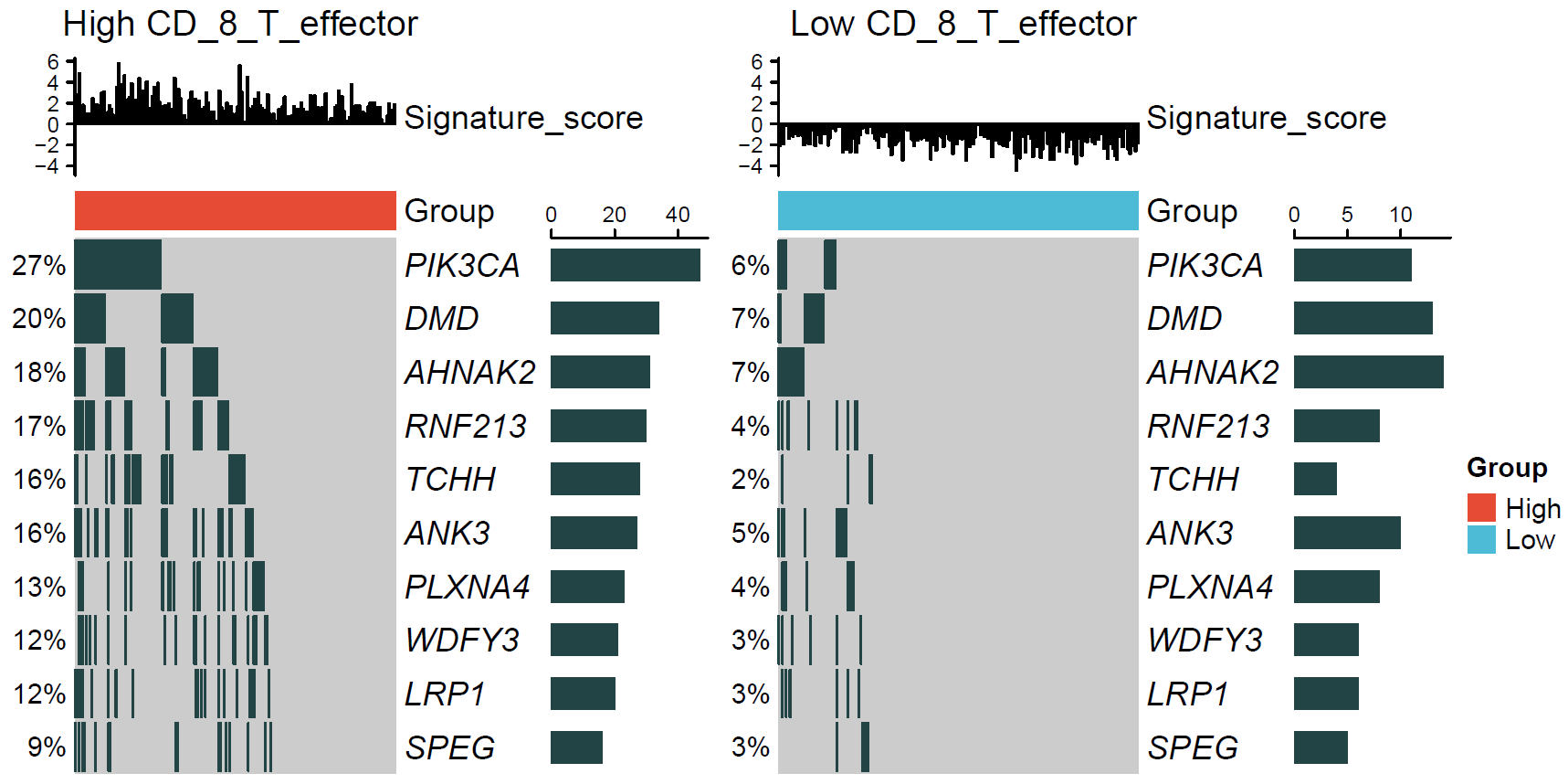
9.6 Other methods to obtaind genomic data
9.6.1 Using TCGAmutations
As its name, the R package TCGAmutations provides pre-compiled, curated somatic mutations from 33 TCGA cohorts along with relevant clinical information for all sequenced samples. You can install it similar to the TCGAbiolinks.
if (!requireNamespace("TCGAmutations", quietly = TRUE)) {
BiocManager::install("PoisonAlien/TCGAmutations")
}It’s quite simple to use:
9.6.2 Using maftools
If you are a user of R package Maftools, you can access and load the data in a similar way (because the author of TCGAmutations and Maftools is the same person).
# The following github can be changed to gitee
# it maybe fast in China mainland
maftools::tcgaAvailable(repo = "github")
maftools::tcgaLoad("STAD", repo = "github")MAF data transformation To prepare the data for the downstream analysis. We need to extract the SNV data in it and transform it into a non-negative matrix.
9.7 References
Wang et al., (2019). The UCSCXenaTools R package: a toolkit for accessing genomics data from UCSC Xena platform, from cancer multi-omics to single-cell RNA-seq. Journal of Open Source Software, 4(40), 1627, https://doi.org/10.21105/joss.01627
Gu, Z. (2022) Complex Heatmap Visualization. iMeta.
Anand Mayakonda et al., (2018) Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Research